Aim:
Calculate the thermodynamical characteristics of amino acids at water-vapor interface
Description:
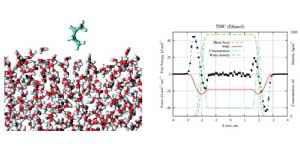
Solvation of 13 neutral amino acid side chain analogs at water –vapor interface was studied by computing high precision free energy profiles of the molecules across the interface using molecular dynamics (MD) simulations. The SPC water model (Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., Hermans, P. A. K. J., Dixon, R., Cornell, W., Fox, T., Chipot, C., Pohorille, A. In: Wilkinson, A., Weiner, P. and van Gunsteren, W. F. editors. Intermolecular Forces, 1981, 3, 331) and OPLS-AA (Jorgensen et al., J Am Chem Soc, 1996, 118, 11225) potential parameter sets were used. A rigorous approach for the computation of high precision free energy profiles at water–vapor interface using constraint force technique is implemented. Methodology of obtaining high precision potential of mean force (PMF) profiles free of simulation artifacts in MD simulations is outlined and discussed. The accuracy of the calculations is examined by comparing the hydration free energies of studied solutes obtained from PMF calculations and separately using Bennett acceptance ratio technique by decoupling solvent-solute interactions in the bulk. All molecules exhibit a free energy minimum at interface. No significant desolvation barrier is observed for any of the studied species. Adsorption energies for studied molecules at water–vapor interface are estimated and compared with experimental observations. We find that for modeled neutral compounds pronounced surface influence on solute solvation vanishes already at 6–7 Å behind the water surface as the solvation free energy approaches the bulk value. The possibility of force field refinement using adsorption free energies is outlined.
Investigators:
Project images:
Project completed:
yes 