Objective
A brief hands-on tutorial on 100ns MD simulation of H3-H4 tetramer and covariance analysis.
Tutorial contents
- System summary
- System Preparation
- Relaxation
- Energy minimization
- NVT ensemble equilibration
- NPT ensemble equilibration, step one
- NPT ensemble equilibration, step two
- Production MD
- Processing the trajectory
- Covariance analysis
- Acknowledgements
System summary
Force field - AMBER99SB-ILDN
System size - 10.092 nm х 10.092 nm х 10.092 nm = 1027.92 nm^3
Number of atoms - 102766.
Number of atoms in H3 histone - 1580
Number of atoms in H4 histone - 1336
Number of water molecules: 32334
Number of Cl ions: 32
Concentration of ions: 0.059 mol/dm^3
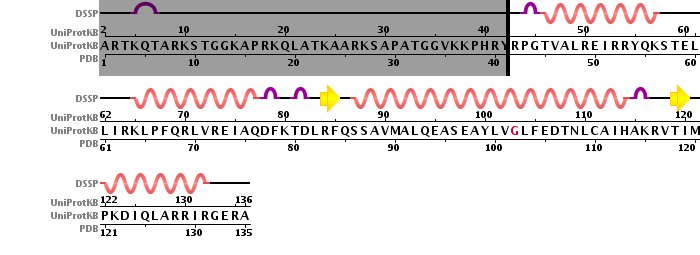
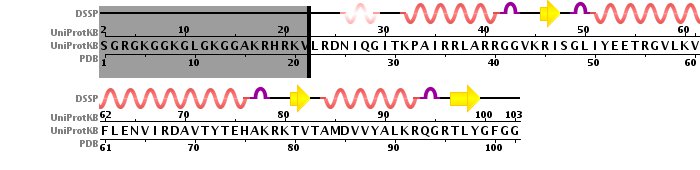
Histone tetramer strucutre was derived from 1KX5 PDB structure, long histone chains were omitted. The pictures below contain sequences with secondary structures. Omitted chains are filled with gray color.
Histone H3 sequence with secondary structure.
Histone H4 sequence with secondary structure.
The resulting structure is provided as "tetramer.pdb" in this archive.
Download and unpack archive in some directory:
wget http://molsim.org/sites/default/files/h3-h3_tetra_tutor.tar tar -xvf h3-h3_tetra_tutor.tar
System Preparation
Step one: generate topology files in Gromacs.
At first we should convert our PDB file into GROMACS native .gro file format:
pdb2gmx_4.5.5d -ter -f tetramer.pdb -o tetramer.gro
-ter - allows to interactively select the terminal capping groups, choose non-polar option.
Choose the AMBER99SB-ILDN force field (number 6) and TIP3P-water model (number 1).
pdb2gmx will show you some useful info:
Now there are 5832 atoms and 350 residues
Total mass in system 40215.772 a.m.u.
Total charge in system 34.000 e
Step two: define box and solvate.
Now we will put our dimer into a periodic box using the following command
editconf_4.5.5d -f tetramer.gro -o tetramer_box.gro -c -d 1.0 -bt cubic
-c centers protein in the periodic cell.
-d 1.0 defines the smallest distance between the protein and the cell boundary.
-bt cubic defines cubical shape of the periodic cell.
system size : 7.968 6.423 6.145 (nm)
diameter : 8.092 (nm)
center : 4.726 9.880 0.692 (nm)
box vectors : 10.595 18.117 10.949 (nm)
box angles : 90.00 90.00 90.00 (degrees)
box volume :2101.66 (nm^3)
shift : 0.320 -4.834 4.354 (nm)
new center : 5.046 5.046 5.046 (nm)
new box vectors : 10.092 10.092 10.092 (nm)
new box angles : 90.00 90.00 90.00 (degrees)
new box volume :1027.92 (nm^3)
Next - adding water to the box
genbox_4.5.5d -cp tetramer_box.gro -cs spc216.gro -p topol.top -o tetramer_solved.gro
-cs spc216.gro determines the water structure file
Output configuration contains 102834 atoms in 32684 residues
Volume : 1027.92 (nm^3)
Density : 1230.32 (g/l)
Number of SOL molecules: 32334
Step three: add ions.
As pdb2gmx said our system got a nonzero total charge: 34 e.
We should add 34 negative ions to make it neutral.
For this step we need an input file for genion: ions.mdp
grompp_4.5.5d -f ions.mdp -c tetramer_solved.gro -p topol.top -o ions.tpr genion_4.5.5d -s ions.tpr -o tetramer_solved_ions.gro -p topol.top -pname NA -nname CL -nn 34
The above line adds ions into the box,
-pname and -nname defines names of positive and negative charges
-nn defines the amount of the negative charges
When prompted, choose group 13 "SOL" as the one which will embed ions.
Picture of the H3-H4 tetramer in water with ions.
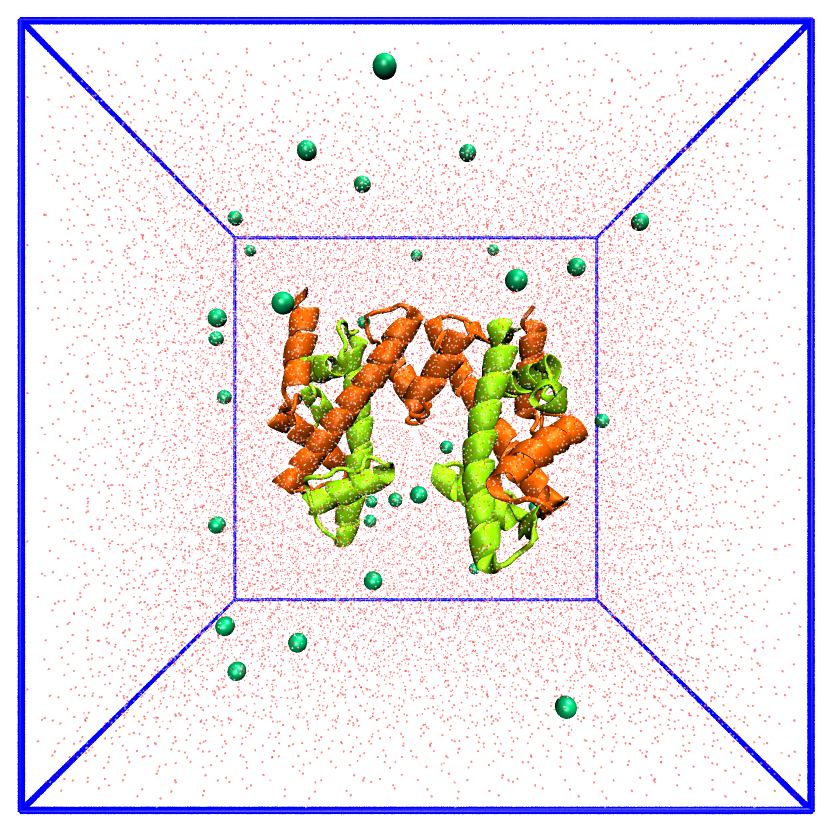
Relaxation
Now our system is ready for relaxation.
The relaxation will be performed in four steps:
1.energy minimization.
grompp_4.5.5d -f minim.mdp -c tetramer_solved_ions.gro -p topol.top -o emin.tpr
file minim.mdp contains the input parameters for energy minimization.
Examples of job submitting is suitable for SLURM job scheduling system and may be used on MSU Lomonosov Supercomputer.
sbatch -n 32 -p regular4 ompi mdrun_4.5.5d_mpi -v -deffnm emin
NOTE: path to your Gromacs suite should be contained in the $PATH environment variable. Or you may use direct link such as '/home/user/software/gromacs/bin/mdrun_mpi'.
Energy plot.
g_energy_4.5.5d -f emin.edr -o potential.xvg
At the prompt, type "10 0" to select Potential (10); zero (0) terminates input.
You can use GNUplot or Xmgrace for visualization, the resulting plot should look something like this.
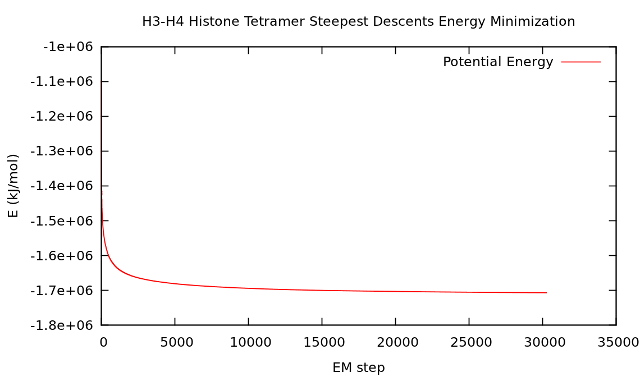
2. NVT ensemble equilibration.
The protein is fixed, only water and ions can move.
grompp_4.5.5d -f nvt.mdp -c emin.gro -p topol.top -o nvt.tpr
file nvt.mdp contains the input parameters for NVT equilibration.
sbatch -n 128 -p regular4 ompi mdrun_4.5.5d_mpi -v -deffnm nvt
Temperature plot.
g_energy_4.5.5d -f nvt.edr -o nvt.xvg
Type "15 0" at the prompt to select the temperature of the system and exit.
The resulting plot should look something like this.
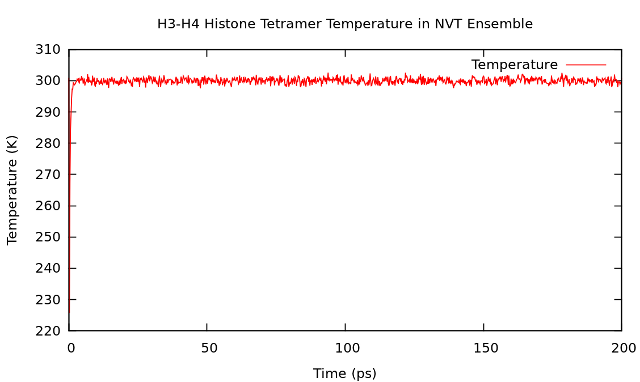
3. NPT ensemble equilibration.
The protein is fixed, only water and ions moves.
grompp_4.5.5d -f npt.mdp -c nvt.gro -t nvt.cpt -p topol.top -o npt.tpr -maxwarn 1
-t key is used to continue the dynamics from the NVT equilibration
-maxwarn key is used to ignore Gromacs warning about position restraining of protein.
sbatch -n 128 -p regular4 ompi mdrun_4.5.5d_mpi -v -deffnm npt
Pressure and density plot.
g_energy_4.5.5d -f npt.edr -o npt.xvg
Type "16 22 0" at the prompt to select the pressure and the density of the system and exit.
The resulting plost should look something like this.
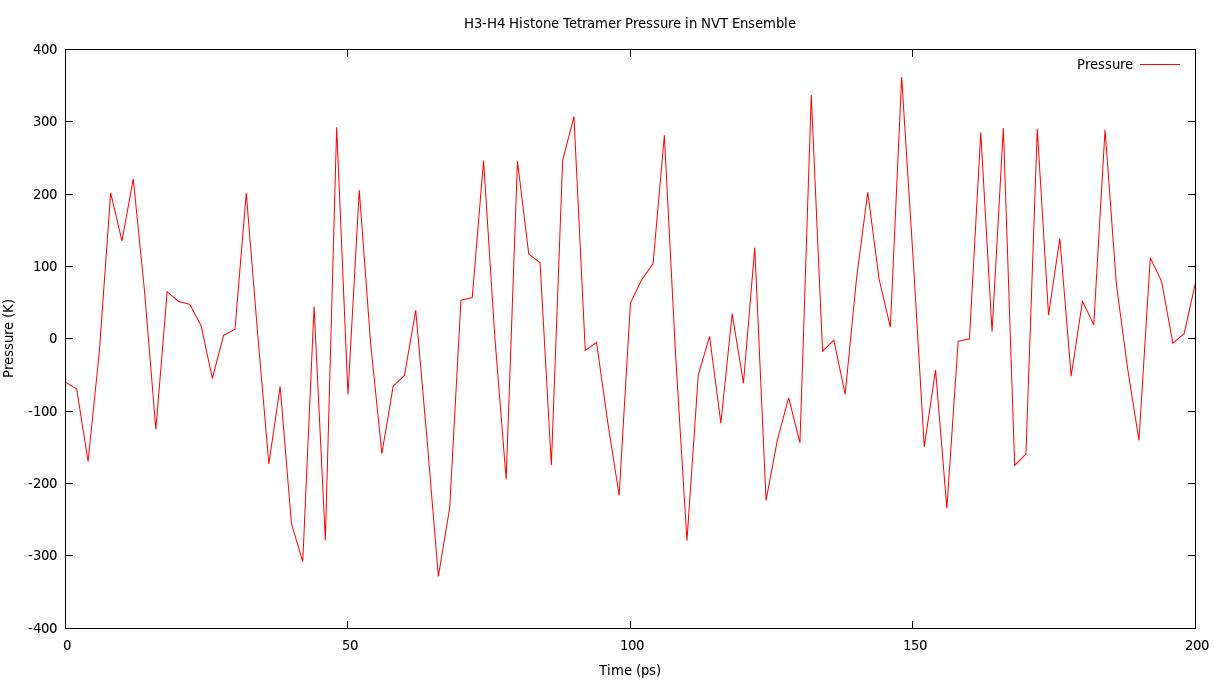
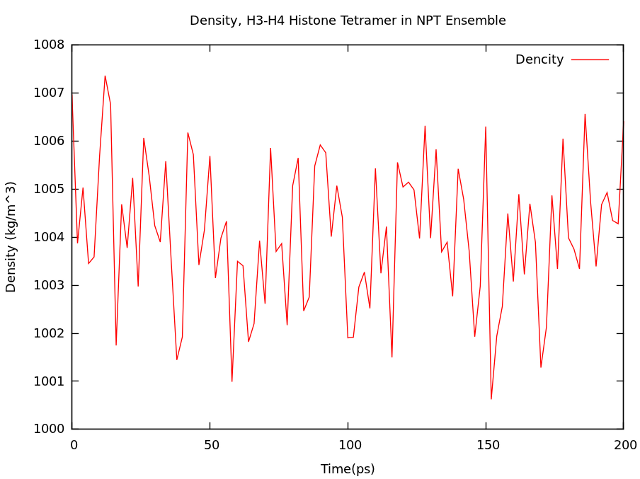
4. NPT ensemble equilibration
The protein is not fixed.
grompp_4.5.5d -f npt1.mdp -c npt.gro -t npt.cpt -p topol.top -o npt1.tpr
sbatch -n 128 -p regular4 ompi mdrun_4.5.5d_mpi -v -deffnm npt1
Production MD
Now we are able to perform dynamics.
grompp_4.5.5d -f md.mdp -c npt1.gro -t npt1.cpt -p topol.top -o md100ns.tpr sbatch -n 256 -p regular4 ompi mdrun_4.5.5d_mpi -v -deffnm md100ns
You can monitor the progress of MD by looking at md100ns.log file.
tail -n 15 nd100ns.log
Processing the trajectory.
When the calculations are completed (about 7 days on 48CPU cluster) we will be able to analyze and visualize the trajectory.
The output trajectory file is about 10 GB, so it may be useful to clean the trajectory and delete the solvent and ions. Due to periodic boundary conditions the protein may walk through the walls of periodic cell. So for the purposes of good visualisation it would be better to process trajectory.
It will be performed in four steps
Step one: put your molecules into the center of the box.
On each step select Protein as group for output and clustering/fitting
trjconv_4.5.5d -f md100ns.trr -o md100nsprot.trr -s npt1.tpr -pbc mol
Step two: dump the first frame from the 'centered' trajectory
trjconv_4.5.5d -f md100nsprot.trr -o refer.pdb -s npt1.tpr -dump 0
Step three: remove jumps using the extracted frame
trjconv_4.5.5d -f md100nsprot.trr -o md100nsnj.trr -s refer.pdb -pbc nojump
Final step: Center the system using the extracted frame as reference, or yse key -fit progressive if your system is too agile.
trjconv_4.5.5d -f md100nsnj.trr -o md100nsfit.trr -s refer.pdb -fit rot+trans
You may use this link to view more instructions in Gromacs documentation.
Now the trajectory is much lighter and can be viewed for example with VMD
But if you use npt1.gro structure file you will lose the information about the protein chains (name of the histones in this situation), use extracted refer.pdb file.
If you want to make a video of MD you may use this set of scripts for VMD.
Covariance analysis
Now we know that the system does not fall apart and it may be interesting to analyse the movements of the histone tetramer using the covariance analysis.
g_covar_4.5.5d -s refer.pdb -f md100nsprot.trr
This will generate the covariance matrix eigenvec.trr and diagonolize it.
It will also generate eigenval.xvg file whith the information about the higest values of eigenvectors.

As you can see the highest values belongs to the firts 5 eigenvectors, and we are able to see movements of the histone tetramer along these eigenvectors.
g_anaeig_4.5.5d -v eigenvec.trr -s refer.pdb -f md100nsprot.trr -first 1 -last 5 -nframes 50 -extr covext.pdb
This will create 5 .pdb files with the trajectories.
Example of the movement along the eigenvector number 2:
The vectors from 2 to 5 represent the collective movements of the core histones with respect to each other. We can filter our MD trajectory to show the movements only along these eigenvectors.
g_anaeig_4.5.5d -s refer.pdb -f md100nsprot.trr -first 2 -last 5 -filt filtprot.pdb
Acknowledgements
The development of tutorial was supported by RFBR grant 12-04-31942.
| Attachment | Size |
|---|---|
| h3-h3_tetra_tutor.tar | 240 KB |
